Introduction
Ce guide s'adresse aux responsables des achats, aux fondateurs de marques et aux responsables de la conformité en charge de l'approvisionnement en dentifrices, bains de bouche et produits blanchissants, et qui sont chargés de l'approbation et de la mise en rayon des produits d'hygiène bucco-dentaire. Les audits et la conformité des produits d'hygiène bucco-dentaire sont les principales causes de retards de lancement, de blocages en douane, de coûts de réétiquetage et d'échecs d'intégration en points de vente, et non la qualité de la formulation.

Soins buccaux visages Les produits d'hygiène bucco-dentaire font l'objet d'un contrôle plus rigoureux que les cosmétiques classiques en raison de leurs ingrédients actifs, des allégations formulées et de la traçabilité des lots. Un blocage en douane de quatre semaines peut immobiliser des sommes importantes correspondant à la quantité minimale de commande. Cet article détaille la logique d'audit par région, les exigences spécifiques aux soins bucco-dentaires et les responsabilités respectives des fabricants d'équipement d'origine (OEM) et des importateurs.
À Oralabx, Nous travaillons spécifiquement avec les marques à ce stade critique, où la formulation est peut-être prête, mais où la mise en œuvre de la conformité détermine si une référence produit parvient réellement sur le marché.
Comment fonctionnent réellement les audits de soins bucco-dentaires (obligatoires ou facultatifs)
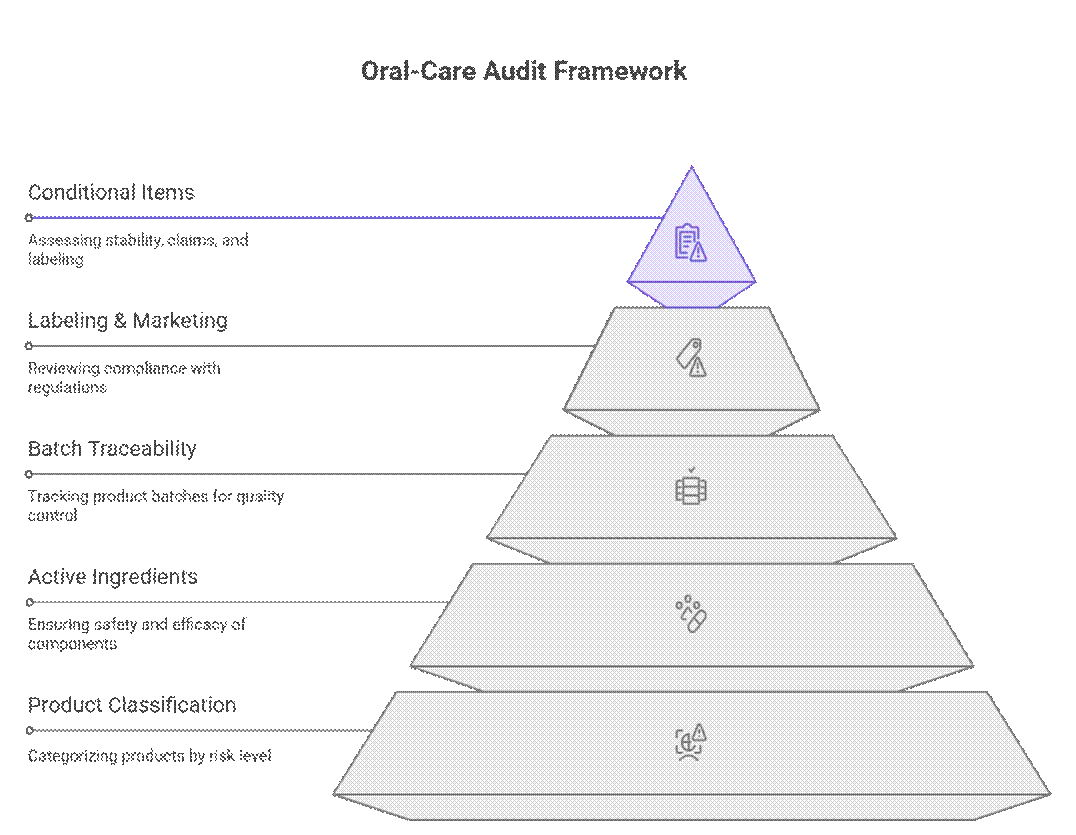
Les organismes de réglementation évaluent la conformité des produits de soins bucco-dentaires selon un cadre hiérarchisé et fondé sur les risques, qui privilégie la classification des produits, les ingrédients actifs et la traçabilité des lots avant l'examen de l'étiquetage ou de la commercialisation. Ce n'est qu'après validation de ces critères qu'ils examinent des éléments conditionnels tels que la durée de stabilité, la justification des allégations ou l'étiquetage transitoire.
Ce que les auditeurs vérifient en premier (non négociable)
Les auditeurs commencent par un ensemble strict d'exigences qui doivent être satisfaites avant que tout examen ultérieur puisse se poursuivre.
- Classification des produits (cosmétiques vs médicaments en vente libre / quasi-médicaments)
La mauvaise classification demeure l'une des erreurs les plus fréquentes dans les exigences d'audit des dentifrices, en particulier lorsque les marques comprennent mal les seuils de classification des produits de soins buccaux (cosmétiques vs OTC) liés aux principes actifs et aux allégations.
- seuils d'ingrédients actifs
Vérifie la conformité aux limites des ingrédients tels que le fluorure ou le CPC, ce qui peut déclencher une surveillance au niveau des médicaments en vente libre ou des médicaments.
- Enregistrement du fabricant et statut GMP
Garantit que l'établissement du fabricant d'équipement d'origine (OEM) est dûment enregistré et fonctionne conformément aux normes BPF approuvées. production de soins bucco-dentaires.
- Traçabilité des lots et préparation aux rappels
Confirme l'existence de dossiers de lots complets permettant de retracer, d'isoler et de rappeler rapidement les produits si les organismes de réglementation ou les détaillants l'exigent.
Qu'est-ce qui est conditionnellement requis ?
Au-delà des éléments non négociables, les auditeurs appliquent une analyse des risques à la documentation et aux tests. L'application de ces règles varie selon le marché et le risque produit, mais les lacunes entraînent souvent des retards ou des approbations conditionnelles.
- Durée de stabilité (accélérée vs. temps réel)
Une stabilité accélérée peut suffire pour les lancements précoces ou à faible risque, mais une stabilité en temps réel est souvent requise pour les produits contenant des principes actifs, ayant une durée de conservation plus longue ou destinés à des marchés plus stricts comme l'UE ou le CCG. Les grands équipementiers mettent en œuvre une stabilité en temps réel en parallèle.
- profondeur de la justification de la revendication
Les allégations cosmétiques de base nécessitent généralement une justification minimale, tandis que les allégations de performance requièrent des données de laboratoire ou cliniques. Les auditeurs signalent les allégations insuffisamment étayées ou non conformes à la formulation finale.
- Langue locale et formats d'étiquetage
Les étiquettes en anglais peuvent convenir pour la vérification, mais l'approbation finale exige généralement un étiquetage conforme dans la langue locale. Les auditeurs signalent fréquemment l'absence de sous-étiquettes, les noms d'ingrédients incorrects ou les traductions d'allégations risquées.
Ce qui est négociable (avec un constructeur automobile de renom)
Bien que la réglementation puisse paraître rigide, les auditeurs expérimentés tiennent compte des réalités pratiques de la production. Avec un constructeur d'origine reconnu et une feuille de route claire en matière de conformité, certaines exigences peuvent être mises en œuvre progressivement ou séquentiellement sans pour autant bloquer l'accès au marché.

- Étiquetage transitoire
Les auditeurs peuvent accepter un étiquetage provisoire pour les premiers tirages si le visuel final est approuvé et programmé. Les grands fabricants d'équipement d'origine (OEM) veillent à la séparation des lots et empêchent la mise en vente d'étiquettes non définitives.
- Documentation progressive lors de la mise à l'échelle
Les auditeurs peuvent accepter un étiquetage provisoire pour les premiers tirages si le visuel final est approuvé et programmé. Les grands fabricants d'équipement d'origine (OEM) veillent à la séparation des lots et empêchent la mise en vente d'étiquettes non définitives.
- Tests en parallèle lors des essais pilotes
Les constructeurs automobiles les plus performants effectuent des tests de stabilité, microbiologiques et d'emballage lors des lots pilotes, ce qui permet de réduire de plusieurs semaines, voire de plusieurs mois, les délais de lancement.
Élément visuel #1 : Hiérarchie des exigences d’audit (produits de soins buccaux)
| Niveau de priorité d'audit | Catégorie d'exigence | Ce que cela comprend | Impact de l'audit en cas de données manquantes |
| Niveau 1 — Non négociable | Classification des produits | Détermination des seuils d'ingrédients actifs par rapport aux médicaments en vente libre/quasi-médicaments | Échec immédiat de l'audit ou blocage en douane |
| État des BPF et des installations | ISO 22716, enregistrement auprès de la FDA, contrôles d'hygiène | Refus d'expédition, disqualification de l'usine | |
| traçabilité des lots | Enregistrements de lots, préparation au rappel, codage des lots | Mesures correctives obligatoires, rappel potentiel | |
| Niveau 2 — Conditionnellement requis | Soutien à la stabilité | Stabilité accélérée et/ou en temps réel liée à la durée de conservation | Approbation conditionnelle, obligation post-commercialisation |
| justification de la réclamation | Données à l'appui des allégations concernant le blanchiment, le fluorure, la plaque dentaire et le CPC | Suppression ou réétiquetage de la réclamation requis | |
| Conformité à l'étiquetage local | Conventions relatives à la langue, au format et à la dénomination des ingrédients | Coûts de réétiquetage, retard de lancement | |
| Niveau 3 — Négociable (selon le fabricant d'origine) | Étiquetage transitoire | Illustrations provisoires avec versions finales approuvées dans les fichiers | Accepté avec un plan de fin de vie documenté |
| Documentation par étapes | Tests à long terme différés pendant la phase de montée en puissance | Accepté si les risques sont évalués et si un calendrier est établi. | |
| Tests en parallèle | Tests de stabilité et microbiologiques lors des essais pilotes | Accepté avec des contrôles de lots rigoureux |
Points clés pour les équipementiers :
Les marques échouent aux audits en considérant les éléments de niveau 2 et 3 comme des conjectures facultatives plutôt que comme un risque maîtrisé. Un constructeur automobile performant contrôle cette hiérarchie de manière proactive.
Éléments d'audit spécifiques aux soins bucco-dentaires : Marques manquantes
La plupart des échecs lors des audits de produits d'hygiène bucco-dentaire ne sont pas dus à l'absence de certificats. Ils surviennent lorsque les marques appliquent des hypothèses cosmétiques générales à des produits plus risqués contenant des ingrédients actifs, des allégations fonctionnelles et destinés à un usage quotidien.

Les auditeurs se concentrent très tôt sur ces détails spécifiques aux soins bucco-dentaires, et les lacunes entraînent souvent des retards, un réétiquetage ou des mesures correctives.
Éléments d'audit spécifiques au dentifrice
Les dentifrices font l'objet de contrôles plus rigoureux en raison des allégations relatives à leurs propriétés actives et thérapeutiques.
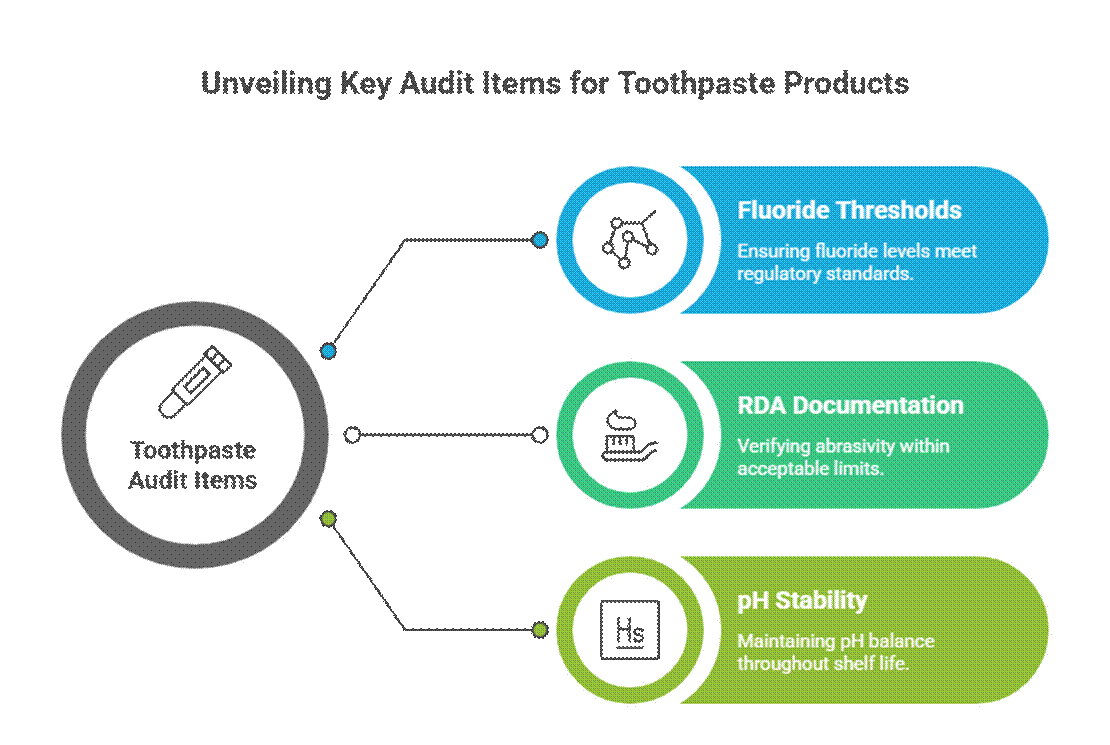
- Seuil de fluorure (pertinence de la monographie de la FDA)
Aux États-Unis, dentifrice au fluor Le dentifrice doit être conforme à la monographie de la FDA relative au fluorure, notamment en ce qui concerne la concentration en principe actif, le format de l'étiquetage et la cohérence des allégations. Les auditeurs exigent également la documentation RDA pour le dentifrice et la validation des tests de stabilité bucco-dentaire pendant toute sa durée de conservation.
- Documentation RDA (limites d'abrasivité)
Les auditeurs s'attendent à ce que les données d'abrasivité relative de la dentine (ARD) confirment la sécurité de l'émail. Des valeurs d'ARD manquantes ou excessives constituent un signal d'alarme courant, notamment pour les produits de blanchiment ou les formulations à base de charbon.
- Stabilité du pH pendant la durée de conservation
Le pH du dentifrice doit rester dans une plage sûre et fonctionnelle tout au long de sa durée de conservation. Les auditeurs examinent les données de stabilité afin de s'assurer que les variations de pH ne compromettent pas l'efficacité du fluorure ni la sécurité bucco-dentaire.
Éléments d'audit spécifiques aux bains de bouche
Audits de conformité des bains de bouche L'accent est fortement mis sur le contrôle microbien et la concentration en principes actifs, notamment pour les formules nécessitant une documentation de conformité aux normes CPC pour les bains de bouche et des tests d'efficacité des conservateurs.
- Classification des boissons sans alcool et des boissons contenant de l'alcool
Les dentifrices font l'objet de contrôles plus rigoureux en raison des allégations relatives à leurs propriétés actives et thérapeutiques.
- Documentation CPC %
Les auditeurs vérifient les limites de concentration et la conformité des allégations pour les formules contenant du CPC.
- Rapports de tests de provocation microbienne
L'absence ou l'obsolescence des tests de validation entraînent souvent des approbations conditionnelles.
- Tests d'efficacité des conservateurs
Les systèmes de conservation défectueux sont considérés comme des risques pour la sécurité des produits.
Signaux d'alarme concernant les produits blanchissants
Les auditeurs examinent de près les allégations réglementaires concernant les dentifrices blanchissants, en particulier les formulations impliquant une modification de l'émail, un blanchiment permanent ou des résultats thérapeutiques pouvant déclencher une surveillance au niveau des médicaments.

- Allégations de blanchiment contre allégations médicamenteuses (États-Unis)
Les allégations impliquant une modification de l'émail ou des résultats thérapeutiques peuvent entraîner une classification du médicament.
- divulgation du peroxyde/non-peroxyde
Les auditeurs exigent une divulgation claire des agents blanchissants et de leurs concentrations.
- Formulation de la revendication qui déclenche un examen réglementaire
Les auditeurs exigent une divulgation claire des agents blanchissants et de leurs concentrations.
Points clés pour les équipementiers :
Les audits des produits de soins bucco-dentaires échouent sur la base des principes actifs et des allégations, et non sur l'esthétique de l'emballage.
Appel à l'action en milieu d'article #1 : Télécharger un exemple de pack de conformité
Examinez la documentation OEM anonymisée et prête pour l'audit avant de vous engager sur une quantité minimale de commande ou de finaliser un lancement sur le marché.
Cadres d'audit régionaux (axés sur l'exécution)
Les audits varient selon les régions, mais la logique reste la même : les autorités réglementaires évaluent d’abord le risque lié au produit, puis la documentation, et enfin l’étiquetage. Ce qui diffère, c’est le lieu où s’exerce le contrôle.
États-Unis — FDA et MoCRA
Aux États-Unis, l'application de la loi se concentre de plus en plus sur les exigences d'audit des soins bucco-dentaires de la FDA, avec Conformité aux normes de soins bucco-dentaires du MoCRA Renforcer la responsabilité des marques de soins bucco-dentaires après leur mise sur le marché.
- Arbre de décision cosmétique vs. médicaments en vente libre
La mauvaise classification est un facteur déclencheur fréquent des mesures coercitives.
- Inscription des installations et liste des produits
Les auditeurs vérifient l'exactitude de l'enregistrement MoCRA et la concordance des références.
- Dossiers de lots et préparation aux événements indésirables
Les marques doivent démontrer la traçabilité de leurs systèmes et leur capacité à traiter les réclamations.
- Allégations blanchissantes : où les marques franchissent la ligne
Les allégations exagérées en matière de blanchiment entraînent souvent un examen minutieux des médicaments.
Union européenne — CPNP et PIF
La conformité aux normes européennes en matière de soins bucco-dentaires via le CPNP et le PIF exige une parfaite adéquation entre la formulation, l'étiquetage et les exigences du CPSR pour les produits de soins bucco-dentaires, la responsabilité incombant à la personne responsable.

- Rôle de la personne responsable (PR)
Le RP doit contrôler et accéder à l'intégralité du fichier d'informations sur le produit.
- attentes en matière d'évaluation de la sécurité
Les CPSR génériques sont fréquemment rejetés pour les produits de soins buccaux.
- Contrôles de stabilité, CPSR et d'étiquetage
Les incohérences entre les documents constituent des points de défaillance courants.
- Exposition à la responsabilité de l'importateur
La responsabilité incombe au RP ou à l'importateur, et non au constructeur d'origine.
Moyen-Orient — GSO 1943
Enregistrement GSO 1943 pour les soins buccaux Cela exerce une pression accrue sur les services douaniers, rendant l'exactitude de l'étiquetage et la cohérence des soumissions essentielles au dédouanement des expéditions.
- délais d'enregistrement des produits
Les dossiers incomplets entraînent souvent la réinitialisation du processus.
- exigences d'étiquetage en arabe
L'absence ou l'inexactitude des sous-étiquettes arabes entraîne fréquemment le blocage des expéditions.
- Causes fréquentes de saisie douanière
Modifications d'étiquettes et incohérences de documentation après soumission.
Micro-cas : Blocage douanier européen dû à une dérive documentaire
Un distributeur européen a importé un dentifrice blanchissant au fluorure produit par un fabricant d'équipement d'origine (OEM) de second rang. Bien que la formule soit conforme, la cargaison a été bloquée en douane en raison d'incohérences entre le dossier d'information produit (DIP), les données de stabilité et les allégations figurant sur l'étiquette finale. Le rapport CPSR faisait référence à une version antérieure de la formule ; les tests de stabilité en temps réel n'avaient pas été effectués et les allégations de blanchiment dépassaient les seuils approuvés par l'évaluateur de sécurité.
Il en a résulté un blocage en douane de quatre semaines, un réétiquetage obligatoire et 11 800 € de frais imprévus, immobilisant ainsi le stock. Si les versions de la formulation, les protocoles de stabilité et les justificatifs des allégations avaient été validés avant la soumission, l’envoi aurait été dédouané sans problème.
C’est pourquoi les fabricants d’équipement d’origine (OEM) expérimentés en soins buccaux effectuent des tests de stabilité en parallèle, figent les versions de formulation avant la finalisation du PIF et réalisent des analyses des risques liés aux allégations avant l’approbation des illustrations, évitant ainsi les dérives de documentation que les auditeurs signalent immédiatement.
Clarification des responsabilités
Une forte conformité aux BPF des fabricants d'équipement d'origine (OEM) de produits de soins buccaux, soutenue par la norme ISO 22716 relative à la fabrication de produits de soins buccaux, garantit que les auditeurs peuvent vérifier les exigences de traçabilité des lots de produits de soins buccaux sans mesures correctives ni rappels.
Chez Oralabx, nous accompagnons les marques en séparant clairement la responsabilité juridique du soutien à l'exécution : nous fournissons une documentation spécifique à chaque lot, prête pour l'audit, tout en signalant les risques de classification et de réclamation avant qu'ils ne deviennent des problèmes d'application de la loi.
Responsabilité de l'importateur vs du fabricant d'équipement d'origine (Section Clarté)
Une séparation claire des responsabilités est essentielle pour réussir les audits de soins bucco-dentaires et éviter toute responsabilité après la mise sur le marché. Les organismes de réglementation n'évaluent pas les intentions ni les accords informels ; ils déterminent qui est légalement responsable et qui contrôle les documents. En cas d'ambiguïté des rôles, la responsabilité du contrôle incombe par défaut à l'importateur, même si le fabricant d'origine a effectué la plupart des tâches de conformité.
Ce dont l'importateur est légalement responsable
Indépendamment du soutien du fabricant d'origine, les autorités réglementaires attribuent la responsabilité finale à la partie qui met le produit sur le marché. Les importateurs ne peuvent se décharger de ces obligations sans un transfert de responsabilité légal explicite.

- Autorisation de mise sur le marché
Obtenir l'approbation légale avant la vente.
- Approbation des réclamations
Toutes les allégations relatives au marketing et à l'emballage relèvent de la responsabilité de l'importateur.
- conservation des documents de vente au détail
Les importateurs doivent conserver et produire sur demande les documents de conformité, notamment l'accès au PIF, les certificats d'analyse, les fiches de données de sécurité et les dossiers de traçabilité des lots.
Ce qu'un constructeur automobile compétent doit fournir
Un fabricant d'équipement d'origine (OEM) qualifié en soins bucco-dentaires soutient la mise en œuvre de la conformité en fournissant une documentation vérifiable et prête pour l'audit, directement liée à chaque lot de production.
- Certification BPF
Actuel, certification BPF reconnue au niveau régional (par exemple, ISO 22716) démontrant des systèmes de fabrication, d'hygiène et de qualité contrôlés.
- Enregistrements de lots et traçabilité
Dossiers complets de fabrication par lots, codage des lots et traçabilité des rappels reliant les matières premières, les cycles de production et les produits finis.
- CoA, FDS, rapports de stabilité et microbiologiques
Certificats d'analyse spécifiques au lot, fiches de données de sécurité, études de stabilité et rapports de tests microbiologiques/de résistance alignés sur la formulation finale.
- Documentation d'exportation
Factures commerciales, listes de colisage, déclarations et documents d'exportation justificatifs précis, conformes aux données et à l'étiquetage des produits enregistrés.
Tableau récapitulatif visuel #2 : Responsabilités de l’importateur et du fabricant d’équipement d’origine (OEM)
Ce tableau précise comment les organismes de réglementation attribuent les responsabilités lors des audits de soins bucco-dentaires et où s'arrête le soutien des fabricants d'équipement d'origine et où commence la responsabilité légale.
Responsabilité de l'importateur par rapport à celle du fabricant d'équipement d'origine (Conformité en matière de soins bucco-dentaires)
| Zone de conformité | Importateur (Responsabilité légale) | OEM (Support à l'exécution) |
| Classification des produits | Décision de classification finale | Entrées de formulation et indicateurs de risque |
| Autorisation de mise sur le marché | Inscription, notification, nomination RP | Préparation des données et des documents |
| Approbation des réclamations | Légalité et justification de la revendication | Examen des risques liés aux réclamations et soutien aux données |
| Conformité aux BPF | Vérifie la qualification OEM | Assure le maintien des systèmes et des audits BPF |
| traçabilité des lots | Conserve et produit des documents | Crée des enregistrements de lots et des codes de lots |
| Données de stabilité et de sécurité | Garantit l'adéquation au marché | Effectue des tests et rédige des rapports |
| Conformité de l'étiquetage | Approbation finale de l'étiquette | Examen des illustrations et conseils de mise en forme |
| Préparation à l'audit du commerce de détail | Conservation des documents | Fourniture et mise à jour des documents |
| Application de la loi et rappels | Responsabilité financière et juridique | soutien aux enquêtes techniques |
Précision importante :
Les constructeurs automobiles facilitent la mise en conformité, mais les importateurs supportent le risque réglementaire, sauf si la responsabilité est contractuellement transférée. Cette distinction est essentielle pour les résultats des audits et l'exposition post-commercialisation.
Ce que fait réellement un bon constructeur automobile lors d'un audit
À RUIQIGO, Les audits ne sont pas considérés comme des événements documentaires. Nos équipes OEM conçoivent les systèmes de fabrication en intégrant dès le départ les points de contrôle réglementaires, en intégrant la traçabilité des lots, le contrôle actif et la vérification de la conformité directement dans la production quotidienne.
Flux de travail de conformité OEM (exécution réelle)
Lors d'un audit, les équipementiers compétents démontrent leur conformité par la maîtrise des processus, et non par une simple documentation réactive. Les auditeurs évaluent si les systèmes sont intégrés aux opérations de fabrication quotidiennes.
- Validation de la formulation sur banc
Les formulations sont validées à l'échelle du laboratoire afin de confirmer les concentrations actives, la plage de pH et la compatibilité des ingrédients avant la mise à l'échelle.
- Stabilité accélérée et en temps réel
L'accélération de la stabilisation permet des soumissions précoces, tandis que des études en temps réel sont menées en parallèle pour valider la durée de conservation et détecter les dérives de formulation.
- Création de feuilles de lot
Chaque cycle de production est documenté par des fiches de lot répertoriant les matières premières, les paramètres de processus, les contrôles en cours de production et les codes de lot.
- Analyses microbiologiques et chimiques
Les produits finis sont soumis à des tests microbiologiques et à une vérification chimique afin de confirmer leur innocuité, l'efficacité de leur conservation et la conformité des ingrédients actifs.
- délivrance du certificat d'authenticité
Des certificats d'analyse spécifiques à chaque lot sont émis et vérifiés par rapport aux spécifications avant la mise sur le marché.
- Documentation d'exportation et douanière
Les documents d'expédition, les déclarations et les dossiers de conformité sont alignés sur les données des produits enregistrés afin d'éviter les divergences douanières.
Comment les fabricants bas de gamme échouent
Les échecs d'audit sont souvent dus à des faiblesses systémiques plutôt qu'à des erreurs isolées. Fabricants de bas niveau Ils manquent généralement d'infrastructures de conformité et ne réagissent que lorsque des problèmes surviennent.
- Traçabilité des lots manquante
Des dossiers de lots inadéquats ou incohérents empêchent des rappels efficaces et sapent immédiatement la confiance des auditeurs.
- Réutilisation des fiches de données de sécurité génériques
Les fichiers SDS recyclés ou non spécifiques à un lot témoignent d'un mauvais contrôle de la formulation et sont fréquemment signalés lors des audits.
- Aucun examen des risques liés aux réclamations
Les fabricants qui n'évaluent pas les risques réglementaires liés à leurs allégations marketing exposent leurs marques à des erreurs de classification et à des mesures coercitives.
- Conformité réactive (et non proactive)
La conformité est traitée comme une demande de documents de dernière minute plutôt que comme un système de fabrication intégré, ce qui entraîne des retards, des mesures correctives et des audits infructueux.
Boîte à emporter OEM :
Les manquements à la conformité sont des défaillances de production, et non des problèmes administratifs. Les auditeurs sanctionnent les systèmes défaillants, et non les dossiers manquants.
Appel à contributions en milieu d'article #2 : Demande de consultation RP / MoCRA
Avant toute soumission réglementaire ou expédition, clarifiez avec un spécialiste de la conformité la responsabilité de l'importateur, les risques liés à la classification des produits et les lacunes en matière de documentation.
Chronologie d'audit visualisée (optionnelle mais fortement recommandée)
Les délais d'audit varient selon les régions, mais les retards surviennent systématiquement aux mêmes étapes : décisions de classification, documentation incomplète et corrections d'étiquetage de dernière minute. Comprendre où le temps est réellement perdu permet aux marques de planifier leurs lancements de manière réaliste et d'éviter les immobilisations de stock.

Espace réservé visuel #3 : Chronologie d’audit (États-Unis vs UE vs Moyen-Orient)
Comparaison des calendriers d'audit des soins buccaux
| Étape d'audit | États-Unis (FDA / MoCRA) | Union européenne (CPNP / PIF) | Moyen-Orient (GSO 1943) |
| Classification des produits | Décision entre cosmétiques et médicaments en vente libre | Classification cosmétique | Classification cosmétique |
| Qualification des installations / des équipementiers | Inscription de l'établissement | Qualification OEM via RP | analyse des données du constructeur |
| Examen de sécurité et de stabilité | Pré-marché limité | CPSR + examen de stabilité | Examen de stabilité |
| Examen des allégations et de l'étiquetage | Modéré | Strict, avant le marché | Strict, avant le marché |
| Inscription/notification | Liste des produits | Notification CPNP | Enregistrement du produit |
| Douanes et dédouanement | Contrôle post-commercialisation | Minimal à la frontière | Application de la loi élevée |
| entrée sur le marché | Entrée anticipée autorisée | Entrée après approbation | Entrée après approbation |
| Contrôle post-commercialisation | Haut | Modéré | Faible à modéré |
Points clés pour les équipementiers :
Le délai de mise sur le marché est réduit grâce à des flux de travail de conformité parallèles, et non en omettant des étapes.
Conclusion (Clôture commerciale)
Le succès à long terme dans le secteur des soins bucco-dentaires repose sur des systèmes structurés, et non sur une gestion administrative réactive. Les marques qui intègrent les audits et la conformité de leurs produits de soins bucco-dentaires à leur processus de fabrication, et non à une simple formalité administrative, préservent leurs marges, leurs délais et la confiance des distributeurs. Pour les marques développant leurs gammes de dentifrices, de bains de bouche ou de produits blanchissants, Oralabx se positionne comme un partenaire de confiance pour la mise en œuvre de la conformité : l’entreprise harmonise les formulations, la documentation et les systèmes de gestion des lots avec les réalités des audits aux États-Unis, dans l’Union européenne et au Moyen-Orient, afin que l’approbation réglementaire se traduise par un accès au marché garanti.
FAQ
Q1. Qui est légalement responsable en cas d'échec d'un audit de soins bucco-dentaires : l'importateur ou le fabricant d'équipement d'origine (OEM) ?
L'importateur est responsable par défaut. Les constructeurs automobiles peuvent faciliter la mise en conformité, mais la responsabilité n'est transférée que si cela est explicitement stipulé dans le contrat et accepté par les autorités réglementaires.
Q2. Les références des sachets individuels de bain de bouche nécessitent-elles une documentation complète de conformité ?
Oui. Les sachets ne sont pas exemptés. Les auditeurs évaluent la formulation, les allégations et les données de sécurité, quel que soit le format de l'emballage.
Q3. Est-il possible d'expédier des échantillons de bain de bouche en sachet individuel sans fiche de données de sécurité ni certificat d'analyse ?
Dans la plupart des cas, non. Les échantillons peuvent être expédiés dans des conditions exceptionnelles, mais les fiches de données de sécurité (FDS) et les certificats d'analyse (CoA) sont généralement exigés par les douanes, notamment pour les produits d'hygiène buccale liquides.
Q4. Combien de temps avant le lancement les travaux de mise en conformité doivent-ils commencer ?
Idéalement, 3 à 6 mois avant le lancement. Les produits contenant des actifs ou revendiquant un effet blanchissant devraient entamer leur planification de conformité encore plus tôt.
Q5. Les fiches produits Amazon nécessitent-elles un certificat d'analyse (CoA) ou une fiche de données de sécurité (SDS)
Amazon demande fréquemment des certificats d'analyse (CoA), des fiches de données de sécurité (SDS) et des documents de conformité lors de l'approbation de l'annonce ou des audits postérieurs à la mise en ligne, en particulier pour les produits de soins bucco-dentaires.
Q6. Quels documents doivent être conservés au niveau du détaillant ?
Les détaillants exigent généralement, sur demande, l'accès aux certificats d'analyse, aux fiches de données de sécurité, aux dossiers de traçabilité des lots et à la preuve de l'autorisation de mise sur le marché.
Q7. Aux États-Unis, les allégations de blanchiment sont-elles considérées comme des allégations relatives aux médicaments ?
Oui. Les allégations impliquant un blanchiment permanent, une modification de l'émail ou des effets thérapeutiques déclenchent souvent un examen rigoureux, comparable à celui des médicaments.
Q8. Un responsable de l'UE peut-il également gérer les marchés du CCG ?
Non. Les marchés du CCG exigent des procédures d'enregistrement et de conformité locales distinctes ; un RP de l'UE ne couvre pas automatiquement les obligations réglementaires du Moyen-Orient.
CTA final : Liste de contrôle de conformité (PDF)
Évitez les retards d'audit, les coûts de réétiquetage et les retenues douanières.
Téléchargez notre liste de vérification de conformité aux soins bucco-dentaires au format PDF ou contactez directement un professionnel de la santé bucco-dentaire. spécialiste de la conformité OEM pour valider votre documentation avant le lancement.