
Introduction
This guide is written for procurement leaders, brand founders, and compliance managers sourcing toothpaste, mouthwash, and whitening products who are responsible for getting oral-care SKUs approved and onto shelves. Oral-care product audits and compliance are the leading causes of launch delays, customs holds, relabeling costs, and failed retail onboarding—not formulation quality.

Oral care faces stricter scrutiny than general cosmetics due to actives, claims, and batch traceability, and a 4-week customs hold can trap six figures of MOQ cash. This article breaks down region-by-region audit logic, oral-care–specific requirements, and clear OEM vs importer responsibilities.
At Oralabx, we work with brands specifically at this failure point—where formulation may be ready, but compliance execution determines whether a SKU actually reaches market.
How Oral-Care Audits Actually Work (Mandatory vs Optional)
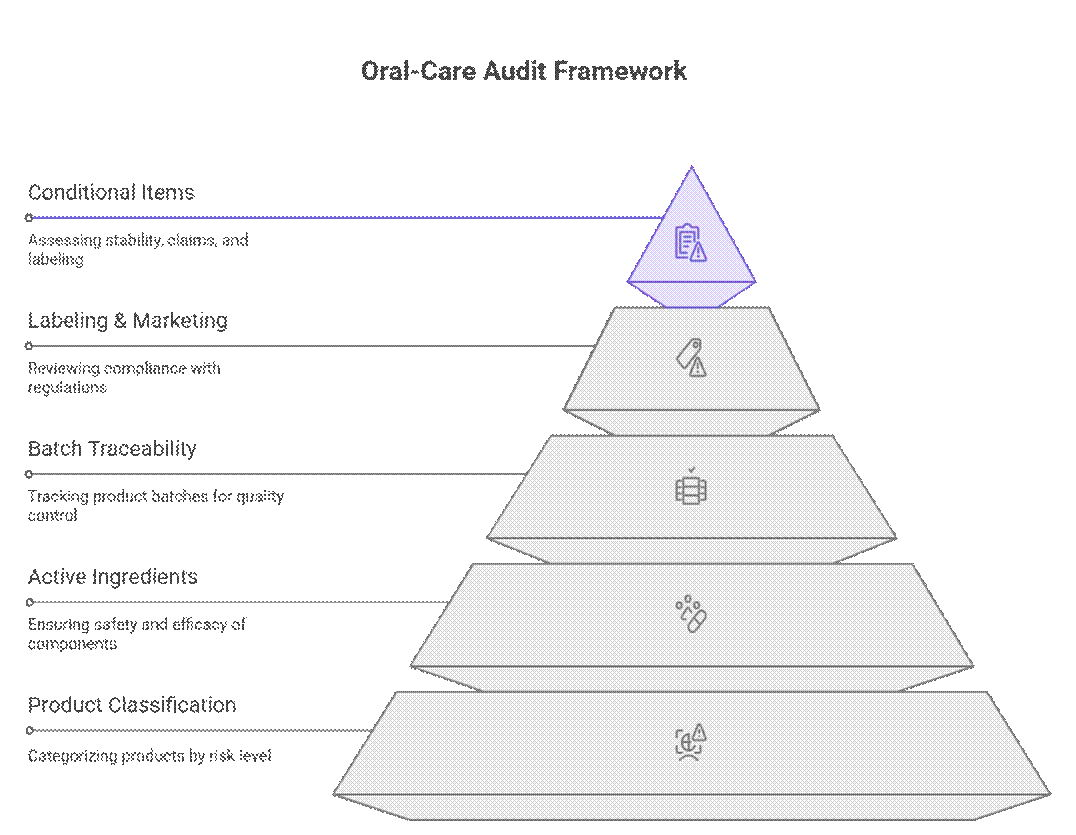
Regulators approach oral-care product audits and compliance using a tiered, risk-based framework that prioritizes product classification, active ingredients, and batch traceability before labeling or marketing review. Only after these pass do they review conditional items like stability duration, claim substantiation, or transitional labeling.
What Auditors Check First (Non-Negotiable)
Auditors begin with a strict set of requirements that must be satisfied before any further review can proceed.
- Product classification (cosmetic vs OTC / quasi-drug)
Misclassification remains one of the most common failures in toothpaste audit requirements, particularly when brands misunderstand oral-care product classification cosmetic vs OTC thresholds tied to actives and claims.
- Active ingredient thresholds
Verifies compliance with limits for ingredients such as fluoride or CPC, which can trigger OTC or drug-level oversight.
- Manufacturer registration & GMP status
Ensures the OEM facility is properly registered and operates under approved GMP standards for oral-care production.
- Batch traceability & recall readiness
Confirms complete batch records exist to trace, isolate, and recall products quickly if required by regulators or retailers.
What is Conditionally Required
Beyond the non-negotiables, auditors apply a risk-based review to documentation and testing. Enforcement varies by market and product risk, but gaps often lead to delays or conditional approvals.
- Stability duration (accelerated vs. real-time)
Accelerated stability may be enough for early or low-risk launches, but real-time stability is often required for products with actives, longer shelf lives, or stricter markets like the EU or GCC. Strong OEMs run real-time stability in parallel.
- Claim substantiation depth
Basic cosmetic claims usually require minimal justification, while performance claims need lab or clinical data. Auditors flag claims that lack evidence or do not match the final formulation.
- Local language and labeling formats
English labels may work for review, but final approval usually requires compliant local-language labeling. Auditors commonly flag missing sub-labels, incorrect ingredient names, or risky claim translations.
What is Negotiable (With a Strong OEM)
While regulations appear rigid, experienced auditors recognize practical manufacturing realities. With a proven OEM and a clear compliance roadmap, some requirements can be phased or sequenced without blocking market entry.

- Transitional labeling
Auditors may accept interim labeling for early runs if the final artwork is approved and scheduled. Strong OEMs enforce batch segregation and prevent non-final labels from entering retail.
- Staged documentation during scale-up
Auditors may accept interim labeling for early runs if the final artwork is approved and scheduled. Strong OEMs enforce batch segregation and prevent non-final labels from entering retail.
- Parallel testing during pilot runs
Strong OEMs run stability, microbial, and packaging tests during pilot batches, cutting weeks or months from launch timelines.
Visual Placeholder #1: Audit Requirement Hierarchy (Oral-Care Products)
| Audit Priority Level | Requirement Category | What This Includes | Audit Impact if Missing |
| Level 1 — Non-Negotiable | Product classification | Cosmetic vs OTC / quasi-drug determination, active ingredient thresholds | Immediate audit failure or customs hold |
| GMP & facility status | ISO 22716, FDA registration, hygiene controls | Shipment rejection, factory disqualification | |
| Batch traceability | Batch records, recall readiness, lot coding | Mandatory corrective action, potential recall | |
| Level 2 — Conditionally Required | Stability support | Accelerated and/or real-time stability tied to shelf life | Conditional approval, post-market obligation |
| Claim substantiation | Data supporting whitening, fluoride, plaque, CPC claims | Claim removal or relabeling required | |
| Local labeling compliance | Language, format, ingredient naming conventions | Relabeling costs, launch delay | |
| Level 3 — Negotiable (OEM-Dependent) | Transitional labeling | Interim artwork with approved final versions on file | Accepted with documented sunset plan |
| Staged documentation | Deferred long-term tests during scale-up | Accepted if risk-assessed and scheduled | |
| Parallel testing | Stability and microbial tests during pilot runs | Accepted with strong batch controls |
OEM takeaway:
Brands fail audits by treating Level 2 and Level 3 items as optional guesswork instead of managed risk. A strong OEM controls this hierarchy proactively.
Oral-Care–Specific Audit Items Brands Miss
Most oral-care audit failures are not caused by missing certificates. They occur when brands apply general cosmetic assumptions to higher-risk products with active ingredients, functional claims, and daily use.

Auditors focus on these oral-care–specific details early, and gaps often lead to delays, relabeling, or corrective actions.
Toothpaste-Specific Audit Items
Toothpaste is audited more aggressively due to active and therapeutic-style claims.
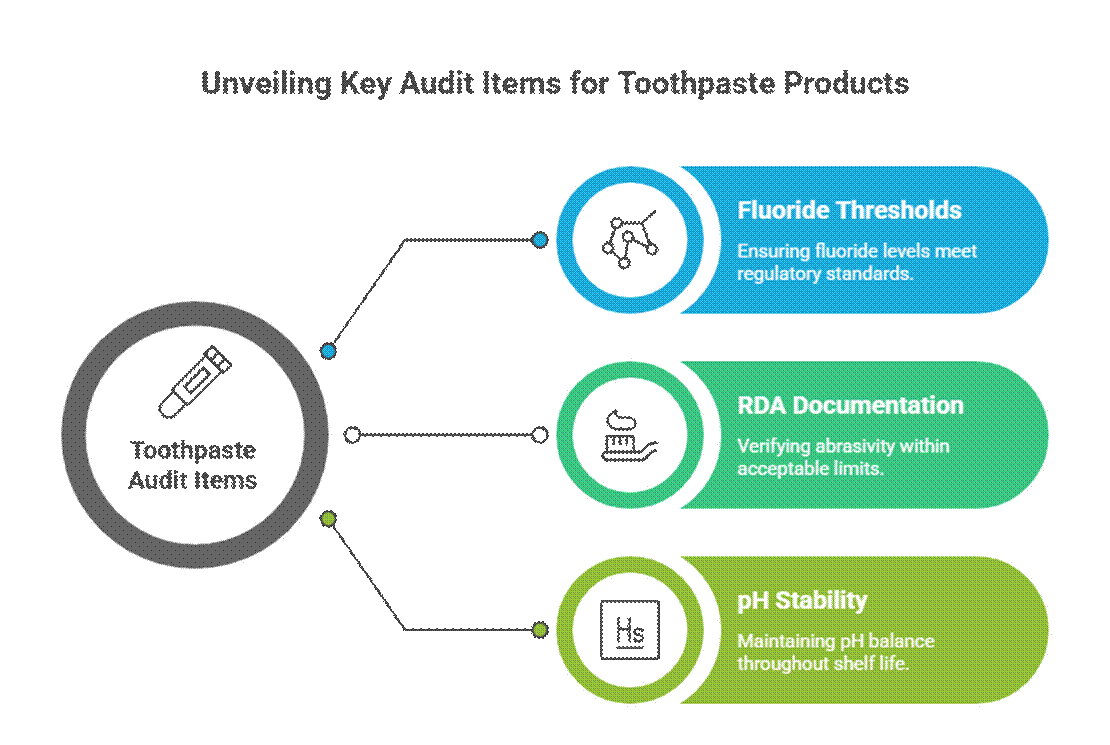
- Fluoride thresholds (FDA monograph relevance)
In the U.S., fluoride toothpaste must comply with the fluoride toothpaste FDA monograph, including active concentration, labeling format, and claim alignment. Auditors also expect RDA documentation for toothpaste and validated oral-care stability testing requirements over the full shelf life.
- RDA documentation (abrasivity limits)
Auditors expect Relative Dentin Abrasivity (RDA) data to confirm enamel safety. Missing or excessive RDA values are a common red flag, especially for whitening or charcoal formulations.
- pH stability over shelf life
Toothpaste pH must remain within a safe, functional range throughout its shelf life. Auditors review stability data to ensure pH drift does not compromise fluoride efficacy or oral safety.
Mouthwash-Specific Audit Items
Mouthwash compliance audits focus heavily on microbial control and active concentration, especially for formulas requiring CPC mouthwash compliance documentation and preservative efficacy testing.
- Alcohol-free vs alcohol-containing classifications
Toothpaste is audited more aggressively due to active and therapeutic-style claims.
- CPC % documentation
Auditors check concentration limits and claim alignment for CPC-containing formulas.
- Microbial challenge test reports
Missing or outdated challenge tests often lead to conditional approvals.
- Preservative efficacy testing
Weak preservative systems are treated as product safety risks.
Whitening Product Red Flags
Auditors closely examine whitening toothpaste regulatory claims, particularly language implying enamel modification, permanent whitening, or therapeutic outcomes that can trigger drug-level oversight.

- Whitening claims vs. drug claims (U.S.)
Claims implying enamel alteration or therapeutic outcomes can trigger drug classification.
- Peroxide / non-peroxide disclosure
Auditors require clear disclosure of whitening actives and concentrations.
- Claim wording that triggers regulatory review
Auditors require clear disclosure of whitening actives and concentrations.
OEM takeaway:
Oral-care audits fail on actives and claims, not packaging aesthetics.
Mid-Article CTA #1: Download Sample Compliance Pack
Review anonymized, audit-ready OEM documentation before committing MOQ or finalizing a market launch.
Regional Audit Frameworks (Execution-Focused)
Audits vary by region, but the logic is consistent: regulators assess product risk first, documentation second, and labeling last. What differs is where enforcement occurs.
United States — FDA & MoCRA
U.S. enforcement increasingly centers on FDA oral-care audit requirements, with MoCRA oral-care compliance expanding post-market accountability for oral-care brands.
- Cosmetic vs. OTC decision tree
Misclassification is a common trigger for enforcement.
- Facility registration & product listing
Auditors verify MoCRA registration accuracy and SKU alignment.
- Batch records & adverse event readiness
Brands must demonstrate traceability and complaint-handling systems.
- Whitening claims: where brands cross the line
Overstated whitening claims often trigger drug scrutiny.
European Union — CPNP & PIF
EU oral-care compliance via CPNP and PIF demands full alignment between formulation, labeling, and CPSR requirements for oral-care products, with liability resting on the Responsible Person.

- Responsible Person (RP) role
The RP must control and access the full Product Information File.
- Safety Assessment expectations
Generic CPSRs are frequently rejected for oral-care products.
- Stability, CPSR, and labeling checks
Inconsistencies across documents are common failure points.
- Importer liability exposure
Liability sits with the RP or importer, not the OEM.
Middle East — GSO 1943
GSO 1943 oral-care registration places enforcement pressure at customs, making labeling accuracy and submission consistency critical to shipment clearance.
- Product registration timelines
Incomplete submissions often reset the process.
- Arabic labeling requirements
Missing or incorrect Arabic sub-labels commonly cause shipment holds.
- Common causes of customs seizure
Post-submission label changes and documentation mismatches.
Micro-Case: EU Customs Hold Caused by Documentation Drift
A European distributor imported a fluoride whitening toothpaste produced by a low-tier OEM. While the formulation itself was compliant, the shipment was stopped at customs due to inconsistencies between the Product Information File (PIF), stability data, and final label claims. The CPSR referenced an earlier formulation version; real-time stability had not been initiated, and whitening claims exceeded what the safety assessor had approved.
The result was a four-week customs hold, mandatory relabeling, and €11,800 in unplanned costs—while inventory capital sat idle. Had formulation versions, stability protocols, and claim substantiation been locked before submission, the shipment would have cleared without intervention.
This is why experienced oral-care OEMs run parallel stability testing, freeze formulation versions before PIF finalization, and perform claim-risk reviews before artwork approval—preventing documentation drift that auditors immediately flag.
Responsibility Clarification
Strong oral-care OEM GMP compliance, supported by ISO 22716 oral-care manufacturing, ensures auditors can verify oral-care batch traceability requirements without corrective actions or recalls.
At Oralabx, we support brands by clearly separating legal liability from execution support—supplying batch-specific, audit-ready documentation while flagging classification and claim risks before they become enforcement issues.
Importer vs OEM Responsibility (Clarity Section)
Clear separation of responsibility is essential to passing oral-care audits and avoiding post-market liability. Regulators do not assess “intent” or informal understandings—they assess who is legally responsible and who controls the documents. When roles are unclear, enforcement defaults to the importer, even if the OEM performed most compliance tasks.
What the Importer is Legally Liable For
Regardless of OEM support, regulators assign ultimate accountability to the party placing the product on the market. Importers cannot outsource these obligations without explicit legal reassignment.

- Market authorization
Ensuring legal approval before sale.
- Claims approval
All marketing and on-pack claims fall under importer liability.
- Retail documentation retention
Importers must retain and produce compliance documents upon request, including PIF access, CoAs, SDS, and batch traceability records.
What a Competent OEM Must Provide
A qualified oral-care OEM supports compliance execution by supplying verifiable, audit-ready documentation tied directly to each production batch.
- GMP certification
Current, region-recognized GMP certification (e.g., ISO 22716) demonstrating controlled manufacturing, hygiene, and quality systems.
- Batch records & traceability
Complete batch manufacturing records, lot coding, and recall traceability linking raw materials, production runs, and finished goods.
- CoA, SDS, stability & microbial reports
Batch-specific Certificates of Analysis, Safety Data Sheets, stability studies, and microbial/challenge test reports aligned to the final formulation.
- Export documentation
Accurate commercial invoices, packing lists, declarations, and supporting export documents consistent with registered product data and labeling.
Visual Placeholder #2: Importer vs OEM Responsibility Table
This table clarifies how regulators assign responsibility during oral-care audits and where OEM support ends, and legal liability begins.
Importer vs OEM Responsibility (Oral-Care Compliance)
| Compliance Area | Importer (Legal Liability) | OEM (Execution Support) |
| Product classification | Final classification decision | Formulation input and risk flags |
| Market authorization | Registration, notification, RP appointment | Data and document preparation |
| Claims approval | Claim legality and substantiation | Claim-risk review and data support |
| GMP compliance | Verifies OEM qualification | Maintains GMP systems and audits |
| Batch traceability | Retains and produces records | Creates batch records and lot coding |
| Stability & safety data | Ensures adequacy for the market | Conducts tests and issues reports |
| Labeling compliance | Final label approval | Artwork review and format guidance |
| Retail audit readiness | Document retention | Document supply and updates |
| Enforcement & recalls | Financial and legal responsibility | Technical investigation support |
Key clarification:
OEMs enable compliance, but importers absorb regulatory risk unless liability is contractually reassigned. This distinction is central to audit outcomes and post-market exposure.
What a GOOD OEM Actually Does During an Audit
At RUIQIGO, audits are not treated as document events. Our OEM teams design manufacturing systems around regulatory checkpoints from day one—embedding batch traceability, active control, and compliance verification directly into daily production.
OEM Compliance Workflow (Real Execution)
During an audit, competent OEMs demonstrate compliance through process control, not reactive paperwork. Auditors assess whether systems are embedded in daily manufacturing operations.
- Bench formulation validation
Formulations are validated at bench scale to confirm active concentrations, pH range, and ingredient compatibility before scale-up.
- Accelerated + real-time stability
Accelerated stability supports early submissions, while real-time studies run in parallel to validate shelf life and detect formulation drift.
- Batch sheet creation
Each production run is supported by batch sheets documenting raw materials, process parameters, in-process controls, and lot codes.
- Microbial & chemical testing
Finished goods undergo microbial testing and chemical verification to confirm safety, preservative performance, and active ingredient compliance.
- CoA issuance
Batch-specific Certificates of Analysis are issued and cross-checked against specifications before release.
- Export & customs documentation
Shipping documents, declarations, and compliance files are aligned with registered product data to prevent customs discrepancies.
How Low-Tier Manufacturers Fail
Audit failures often trace back to systemic weaknesses rather than isolated mistakes. Low-tier manufacturers typically lack compliance infrastructure and respond only when issues surface.
- Missing batch traceability
Inadequate or inconsistent batch records prevent effective recalls and immediately undermine auditor confidence.
- Generic SDS reuse
Recycled or non–batch-specific SDS files signal poor formulation control and are frequently flagged during audits.
- No claim-risk review
Manufacturers that do not assess regulatory risk tied to marketing claims expose brands to misclassification and enforcement action.
- Reactive (not proactive) compliance
Compliance is treated as a last-minute document request rather than an integrated manufacturing system, leading to delays, corrective actions, and failed audits.
OEM Takeaway Box:
Compliance failures are manufacturing failures, not paperwork issues. Auditors penalize weak systems, not missing files.
Mid-Article CTA #2: Request RP / MoCRA Consultation Call
Clarify importer liability, product classification risks, and documentation gaps with a compliance specialist before regulatory submission or shipment.
Visualized Audit Timeline (Optional but Strong)
Audit timelines vary by region, but delays consistently occur at the same execution points—classification decisions, incomplete documentation, and late-stage labeling corrections. Understanding where time is actually lost allows brands to plan launches realistically and avoid inventory lockups.

Visual Placeholder #3: Audit Timeline Chart (U.S. vs EU vs Middle East)
Oral-Care Audit Timeline Comparison
| Audit Stage | United States (FDA / MoCRA) | European Union (CPNP / PIF) | Middle East (GSO 1943) |
| Product classification | Cosmetic vs OTC decision | Cosmetic classification | Cosmetic classification |
| Facility / OEM qualification | Facility registration | OEM qualification via RP | OEM data review |
| Safety & stability review | Limited pre-market | CPSR + stability review | Stability review |
| Claims & labeling review | Moderate | Strict, pre-market | Strict, pre-market |
| Registration/notification | Product listing | CPNP notification | Product registration |
| Customs & clearance | Post-market enforcement | Minimal at the border | High enforcement |
| Market entry | Early entry allowed | Entry after approval | Entry after approval |
| Post-market enforcement | High | Moderate | Low-Moderate |
OEM takeaway:
Time-to-market is compressed by parallel compliance workflows, not by skipping steps.
Conclusion (Commercial Close)
Long-term success in oral care depends on structured systems, not reactive paperwork. Brands that treat oral-care product audits and compliance as a manufacturing discipline, not a submission task, protect margins, timelines, and retailer trust. For brands scaling toothpaste, mouthwash, or whitening SKUs, Oralabx operates as a compliance execution partner—aligning formulation, documentation, and batch systems to audit realities across the U.S., EU, and Middle East so regulatory approval converts into predictable market access.
FAQs
Q1. Who is legally liable if an oral-care audit fails — importer or OEM?
The importer is legally liable by default. OEMs can support compliance execution, but liability only shifts if explicitly reassigned by contract and accepted by regulators.
Q2. Do single sachet packet mouthwash SKUs require full compliance documentation?
Yes. Sachet formats are not exempt. Auditors assess formulation, claims, and safety data regardless of pack size.
Q3. Can single sachet packet mouthwash samples ship without SDS or CoA?
In most cases, no. Samples may move under limited exceptions, but SDS and CoA are commonly required at customs, especially for liquid oral-care products.
Q4. How long before launch should compliance work start?
Ideally, 3–6 months before launch. Products with actives or whitening claims should start compliance planning even earlier.
Q5. Do Amazon listings require CoA or SDS?
Amazon frequently requests CoAs, SDS, and compliance documents during listing approval or post-listing audits, particularly for oral-care products.
Q6. What documents must be kept at the retailer level?
Retailers typically require access to CoAs, SDS, batch traceability records, and proof of market authorization upon request.
Q7. Do whitening claims count as drug claims in the U.S.?
They can. Claims implying permanent whitening, enamel alteration, or therapeutic effects often trigger drug-level scrutiny.
Q8. Can an EU Responsible Person also handle GCC markets?
No. GCC markets require separate local registration and compliance processes; an EU RP does not automatically cover Middle East regulatory obligations.
Final CTA: Compliance Checklist PDF
Avoid audit delays, relabeling costs, and customs holds.
Download our Oral-Care Compliance Checklist PDF or speak directly with an OEM compliance specialist to validate your documentation before launch.





